食品藥品監督管理局(FDA)主管:食品、藥品(包括獸藥)、醫療器械、食品添加劑、化妝品、動物食品及藥品、酒精含量低於7%的葡萄酒飲料以及電子產品的監督檢驗;產品在使用或消費過程中產生的離子、非離子輻射影響人類健康和安全項目的測試、檢驗和出證。根據規定,上述產品必須經過FDA檢驗證明安全後,方可在市場上銷售。FDA有權對生產廠家進行視察、有權對違法者提出起訴。
食品安全和實用營養中心(CFSAN)
該中心是FDA工作量最大的部門。它負責除了美國農業部管轄的肉類、家禽及蛋類以外的全美國的食品安全。食品安全和營養中心致力於減少食源性疾病,促進食品安全。並促進各種計劃,如:HACCP計劃的推廣實施等。
該中心的職能包括:確保在食品中添加的物質及色素的安全;確保通過生物工藝開發的食品和配料的安全;負責在正確標識食品(如成分、營養健康聲明)和化妝品方面的管理活動;制定相應的政策和法規,以管理膳食補充劑、嬰兒食物配方和醫療食品;確保化妝品成分及產品的安全,確保正確標識;監督和規範食品行業的售後行為;進行消費者教育和行為拓展;與州和地方政府的合作項目;協調國際食品標準和安全等。
藥品評估和研究中心(CDER)
該中心旨在確保處方藥和非處方藥的安全和有效,在新藥上市前對其進行評估,並監督市場上銷售的一萬余種藥品以確保產品滿足不斷更新的最高標準。同時,該中心還監管電視、廣播以及出版物上的藥品的廣告的真實性。嚴格監管藥品,提供給消費者準確安全的信息。
設備安全和放射線保護健康中心(CDRH)
該中心在確保新上市的醫療器械的安全和有效。因為在世界各地有兩萬多家企業生產從血糖監測儀到人工心臟瓣膜等超過八萬種各種類型的醫療器械。這些產品都是同人的生命息息相關的,因而該中心同時還監管全國範圍內的售後服務等。對於一些象微波爐、電視機、移動電話等能產生放射線的產品,該中心也確定了一些相應的安全標準。
生物製品評估和研究中心(CBER)
該中心監管那些能夠預防和治療疾病的生物製品,因此比化學綜合性藥物更加複雜,它包括對血液、血漿、疫苗等的安全性和有效性進行科學研究。
獸用藥品中心(CVM)
該中心監管動物的食品及藥品,以確保這些產品在維持生命,減輕痛苦等方面的實用性、安全性和有效性。美國食品藥品監督管理局控制瘋牛病的工作也是通過獸藥中心對飼料製造商的檢查得以施行。 2007年12月19日,美國食品藥品監督管理局宣佈建立一個用以跟蹤食物系統中的克隆動物的數據庫,借以使相關鑒別程序得以有效進行。這個數據庫將成為全國動物識別系統(National Animal Identification System)的一部分,該系統用於跟蹤全美所有從還在農場飼養到已上餐桌的家畜。
按照《美國第107-188公共法》 必須向FDA登記的外國食品生產加工企業如下:
1、 酒和含酒類飲料;
2、 嬰兒及兒童食品;
3、 麵包糕點類;
4、 飲料;
5、 糖果類(包括口香糖);
6、 麥片和即食麥片類;
7、 奶酪和奶酪製品;
8、 巧克力和可可類食品;
9、 咖啡和茶葉產品;
10、 食品用色素;
11、 減肥常規食品和藥用食品、肉替代品;
12、 補充食品(即國內的健康食品、維生素類藥品以及中草藥製品);
13、 調味品;
14、 魚類和海產品;
15、 往食品里置放和直接與食品接觸的材料物質及製品;
16、 食品添加劑和安全的配料類食用品;
17、 食品代糖;
18、 水果和水果產品;
19、 食用膠、乳酶、布丁和餡;
20、 冰激淋和相關食品;
21、 仿奶製品;
22、 通心粉和麵條;
23、 肉、肉製品和家禽產品;
24、 奶、黃油和乾奶製品;
25、 正餐食品和鹵汁、醬類和特色製品;
26、 乾果和果仁;
27、 帶殼蛋和蛋製品;
28、 點心(麵粉、肉和蔬菜類);
29、 辣椒、特味品和鹽等;
30、 湯類;
31、 軟飲料和罐裝水;
32、 蔬菜和蔬菜製品;
33、 菜油(包括橄欖油);
34、 蔬菜蛋白產品(方肉類食品);
35、 全麥食品和麵粉加工的食品、澱粉等;
36、 主要或全部供人食用的產品;
醫療器械FDA認證
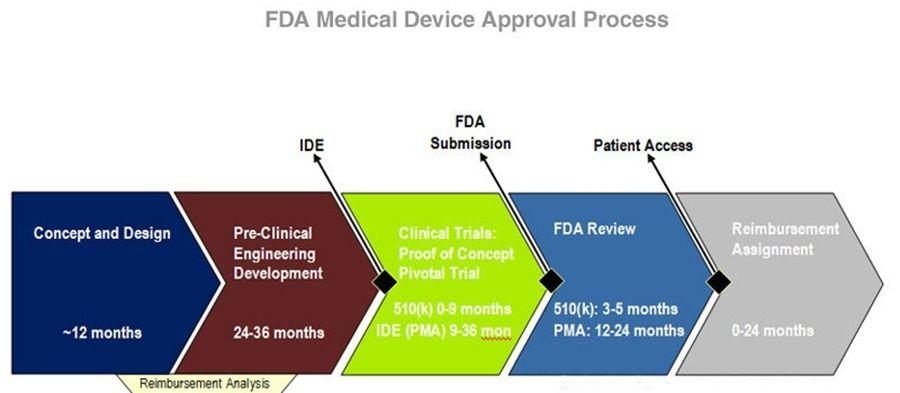
FDA對醫療器械的管理通過器械與放射健康中心(CDRH)進行的,中心監督醫療器械的生產、包裝、經銷商遵守法律下進行經營活動。醫療器械範圍很廣,小到醫用手套,大至心臟起博器,均在FDA監督之下,根據醫療用途和對人體可能的傷害,FDA將醫療器械分為Ⅰ、Ⅱ、Ⅲ類,越高類別監督越多.
如果產品是市場上不曾存在的新穎發明,FDA要求廠家進行嚴格的人體實驗,並有令人信服的醫學與統計學證據說明產品的有效性和安全性。
醫療器械的FDA認證,包括:廠家在FDA註冊、產品的FDA登記、產品上市登記(510表登記)、產品上市審核批准(PMA審核) 醫療保健器械的標籤與技術改造、通關、登記、上市前報告,須提交以下材料:
(1)包裝完整的產成品五份,
(2)器械構造圖及其文字說明,
(3)器械的性能及工作原理;
(4)器械的安全性論證或試驗材料,
(5)製造工藝簡介,
(6)臨床試驗總結,
(7)產品說明書. 如該器械具有放射性能或釋放放射性物質,必須詳細描述.
醫療器械的工廠和產品註冊
FDA對醫療器械有明確和嚴格的定義,其定義如下:「所謂醫療器械是指符合以下條件之儀器、裝置、工具、機械、器具、插入管、體外試劑及其它相關物品,包括組件、零件或附件:明確列於National Formulary或the Unite States Pharmacopeia或前述兩者的附錄中者;預期使用於動物或人類疾病,或其它身體狀況之診斷,或用於疾病之治癒、減緩與治療者;預期影響動物或人體身體功能或結構,但不經由新陳代謝來達到其主要目的者」。
只有符合以上定義的產品方被看作醫療器械,在此定義下,不僅醫院內各種儀器與工具,即使連消費者可在一般商店購買之眼鏡框、眼鏡片、牙刷與按摩器等健身器材等都屬於FDA之管理範圍。它與國內對醫療器械的認定稍有不同。
根據風險等級的不同,FDA將醫療器械分為三類(Ⅰ,Ⅱ,Ⅲ),Ⅲ類風險等級最高。FDA將每一種醫療器械都明確規定其產品分類和管理要求,。任何一種醫療器械想要進入美國市場,必須首先弄清申請上市產品分類和管理要求。
FDA針對醫療器械制訂了許多法案,並不時地進行修改和補充,但根本的法案並不多,主要包括:聯邦食品、藥品與化妝品法案(FD&C Act,根本法案);公眾健康服務法案;公正包裝和標識法案;健康和安全輻射控制法案;安全醫療器械法案;現代化法案。對這些法案,FDA給予了非常詳細的解釋,並配套有具體的操作要求。企業在計劃進入美國市場前,需仔細評估針對自己產品相關的法規和具體要求(包括不同的美國產品標準要求)。
在明確了以上信息後,企業就可以著手準備有關的申報資料,並按一定程序向FDA申報以獲取批准認可。對於任何產品,企業都需進行企業註冊(Registration)和產品列名(Listing)。對Ⅰ類產品(佔47%左右),實行的是一般控制(General Control),絕大部分產品只需進行註冊、列名和實施GMP規範,產品即可進入美國市場(其中極少數產品連GMP也豁免,極少數保留產品則需向FDA遞交510(K)申請即PMN(Premarket Notification));對Ⅱ類產品(佔46%左右),實行的是特殊控制(Special Control),企業在進行註冊和列名後,還需實施GMP和遞交510(K)申請(極少產品是510(K)豁免);對Ⅲ類產品(佔7%左右),實施的是上市前許可,企業在進行註冊和列名後,須實施GMP並向FDA遞交PMA(Premarket Application)申請(部分Ⅲ類產品還是PMN)。
對Ⅰ類產品,企業向FDA遞交相關資料後,FDA只進行公告,並無相關證件發給企業;對Ⅱ、Ⅲ類器械,企業須遞交PMN或PMA,FDA在公告的同時,會給企業以正式的市場准入批准函件(Clearance),即允許企業以自己的名義在美國醫療器械市場上直接銷售其產品。至於申請過程中是否到企業進行現場GMP考核,則由FDA根據產品風險等級、管理要求和市場反饋等綜合因素決定。